Prehospital TBI - Beyond the "Code"
/Of the injuries that one will care for in the pre-hospital setting, traumatic brain injury is one of the most challenging. Quite often, more than one organ system has been injured and they require rapid, thoughtful, and precise management of their airway and hemodynamics. In addition, TBI patients require frequent reassessment to detect progression of the primary neurologic injury. This is easier said than done in the dynamic, unpredictable, and resource-limited prehospital environment.
To help simplify their care, the following “Code of Care” forms the core principles that characterize optimal TBI care:
- NO Hypoxia (SpO2 < 90%) – therefore, apneic oxygenation for all TBI patients
- NO Hypotension (sBP < 90 mmHg) – greatest iatrogenic risk is with induction and provision of positive pressure ventilation
- Blown pupil -> Hyperosmotic therapy + Hyperventilate
To provide truly excellent care, we must go beyond the basic “Code of Care” and add a few more “commandments”:
- If you are only giving hyperosmolar therapy (HOT, i.e. Mannitol or Hypertonic Saline (HTS)) for anisocoria or extensor posturing, then you aren’t giving it enough.
- NO HYPERoxia
- No HYPOor HYPER carbia
- A normal ETCO2, does NOT mean the patient is NORMOcarbic
- If using ketamine for induction or sedation, it should NOT be used in isolation
- Hypotension isn’t <90 mmHg. It’s < 110 mmHg.
- CPP optimization is a core component of optimal ICP management. If deterioration is occurring, assume ICP is 20 mmHg and ensure the MAP is ≥80 mmHg.
Let’s elaborate...
Have a Low Threshold for Giving HOT
Anisocoria and extensor posturing are both well-recognized clinical manifestations of progressive mass effect and cerebral herniation. But the most deadly herniation may not produce either of these findings. For example, a posterior fossa hemorrhage is going to cause tonsillar herniation, producing tetraplegia with small fixed pupils. Subfalcine herniation will cause their level of arousal to be depressed without seeing any posturing, anisocoria, or pupillary dilation. Untreated subfalcine herniation results in obstructed blood flow through the anterior cerebral artery causing infarction, which then extends the primary injury resulting in worsening swelling, leading to further downward herniation.
In addition to anisocoria or extensor posturing, the following should trigger you to give HOT:
- In those with an initial GCS of <13, any drop in the GCS by 2 or more points
- Pupils of any size that are not reactive
- A motor exam with NO response
“But, that will make me over-treat with HOT!” Yes, of course it will. And that’s okay. The issue of just giving HOT has been studied (1,2,3).
Summary
- There is a trend toward clinical benefit in the hypotensive (sBP < 100 mmHg) group.
- There is a signal of physiologic benefit (lower ICP, higher CPP) in normotensive patients.
- Clinical outcomes do not appear to be harmed with treatment.
Do not give HOT to ALL severe TBI patients. If the clinical situation makes you suspect that your patient is deteriorating, herniating, or their exam is the result of elevated intracranial pressure, just give it.
NO HYPERoxia
Except for pre-oxygenation for intubation. Otherwise, spin that dial down and get them to be NORMOxic. Why? Because Dan Davis (4) and Fred Rincon (5) have demonstrated worsened 30 day survival for those with hyperemia (See figure 1 below).

No HYPO or HYPERcarbia
Dr. Jason McMullan was the first one to teach me about the tremendous importance of CO2 management in those with brain injuries.
1 mmHg change in PaCO2 = 2-4% change in Cerebral Blood Flow
In states of relative ischemia, such as when brain tissue is being compressed, or is markedly edematous leading to poor intracranial compliance, small changes in blood flow are liable to lead to infarction or further herniation, respectively. Here are some clinical observations to back this up. (6, 7, 8):
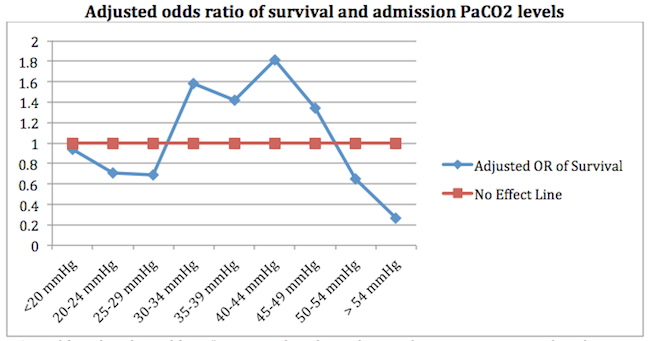
An even more important observation --> correcting your PaCO2 to 35-45 mmHg is associated with improved outcomes (9):
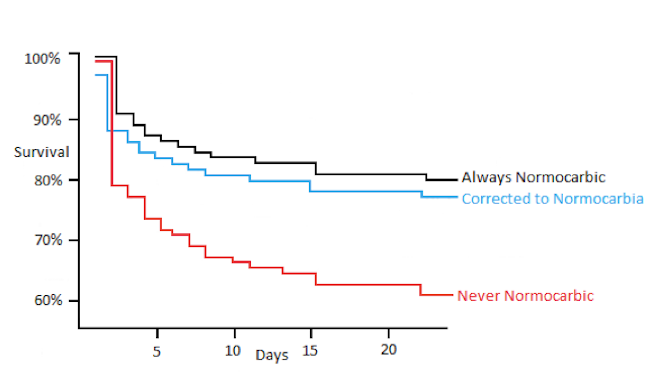
A Normal ETCO2, Does NOT Always Mean that your Patient is NORMOcarbic (10)
Isolated TBI patients --> ETCO2 is accurate. USE IT!
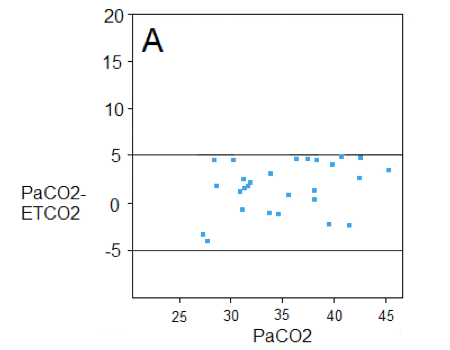
Take home from Figure A-> When your patient is suffering from an isolated TBI, with stable hemodynamics, lacking metabolic derangements, not suffering from thoracic trauma, and is without heart failure or pulmonary disease, your ETCO2 appears to correlate quite well with the PaCO2.
TBI + Acidosis, Chest Trauma, or Hypotension --> ETCO2 is NOT Accurate
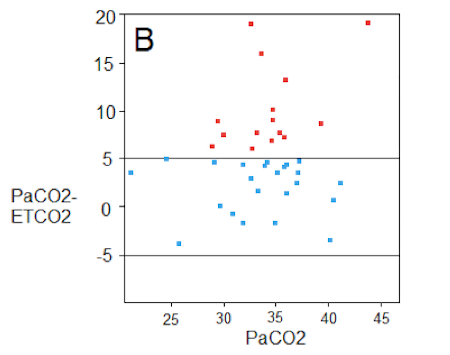
Take home from Figure B - The relationship between ETCO2 and PaCO2 seen in “Figure B” is also reported in those with CHF, Chronic Pulmonary Disease, PE, and other diseases that cause a V/Q mismatch. The ETCO2 appears liable to be significantly discrepant under any of there circumstances. Interpret it with caution.
When using Ketamine for Induction or Sedation, it Should not be Used in Isolation
Ketamine is hot right now. Real hot. For good reason. But the endorsement of its safety might have gone just a bit too far. Back in the 1970s there were several anesthesia-based studies demonstrating that the ICP increases when Ketamine is administered to patients with CSF outflow obstruction. Unfortunately, the increase wasn’t small. It jumped up to 60- 80 mmHg. That is never good. Hence it was kicked to the curb for a while.
Since then some wise folks came along and said “Look, Ketamine preserves airway reflexes, maintains CPP, is an analgesic, doesn’t inhibit bowel motility, reduces vasopressor needs, and might prevent these detrimental spreading depolarizations that everyone is talking about.” So it got studied. BUT NOT AS A MONOTHERAPY. Instead, it has been given with a barbiturate, benzodiazepine, or propofol. It is under those circumstances that the ICP stabilizes or decreases, while there is an improvement in the CPP. There is nothing to say that a slug of ketamine by itself is safe. So, use Ketamine, but only in combination with a benzo or propofol.
Hypotension isn’t <90 mmHg. It’s < 110 mmHg.
The definition of hypotension is certainly a moving target. As a clinician you know to get nervous when you see a SBP of 102 mmHg, but most datasets haven’t had the numbers to detect statistical breakpoints at sBPs above 90 mmHg. Recently, this has been challenged (11):
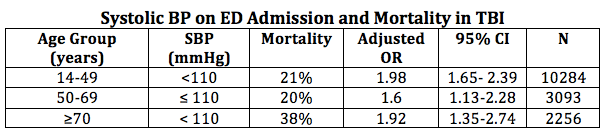
Consider keeping that sBP > 110 mmHg. This should not be interpreted as suggesting that you should start using pressors at a SBP of 108 mmHg. Rather, hypotension should be avoided by providing volume resuscitation early in those you suspect of having hypovolemia and using agents that cause hypotension judiciously. Always remember that hypotension frequently worsens with positive pressure ventilation, notwithstanding the direct cardiovascular effects of the sedation, so anticipate, be prepared, and mitigate.
Optimal ICP management includes ensuring that the MAP is ≥80 mmHg
Whenever optimizing ICP treatment, an adequate MAP must be achieved. You all remember our auto-regulation curve? (12)
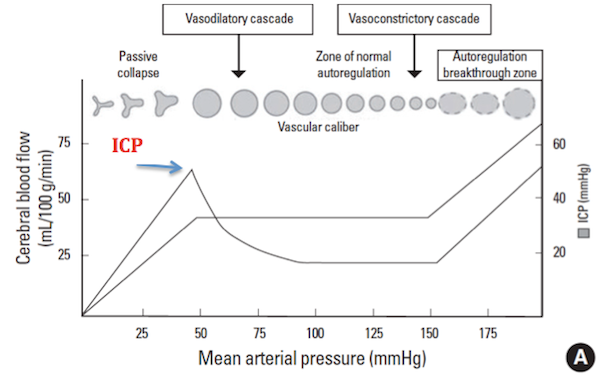
This figure illustrates that the ICP cannot be expected to be lowest at MAPs of < 80 mmHg as a result of the cerebral blood flow preserving cerebral vasodilation that occurs at MAPs below this target. As the MAP increases, there is a reduction in blood vessel diameter, and therefore cerebral blood volume with a preservation of cerebral blood flow. The less blood volume, the lower the intracranial pressure.
Cerebral perfusion pressure (CPP) equals the MAP- ICP. In addition to the ICP optimization that occurs at CPPs of 60-70 mmHg, this level has also been found to be associated with the best clinical outcomes in TBI.
Unfortunately, not all TBI patients retain their auto-regulation during the hyper-acute period. Shooting beyond this in a patient with impaired auto-regulation can worsen the situation:
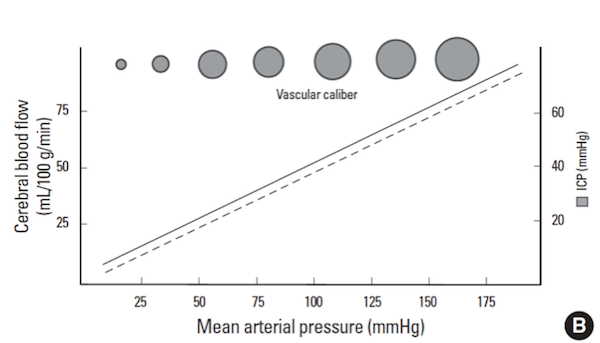
Since there is unlikely to be an ICP lower benefit at MAPs much higher than 80 mmHg, interventions should not imprecisely raise the blood pressure. As shown in the setting of impaired auto-regulation, this can lead to a pathologic rises in ICP.
Bottom line: Assuming an ICP of 20 mmHg, a MAP ~ 80 mmHg is considered to be a solid empiric target until a true ICP can be measured and auto-regulatory curve can be generated.
References
- Wade, et al. Individual Patient Cohort Analysis of the Efficacy of Hypertonic Saline/Dextran in Patients with Traumatic Brain Injury and Hypotension. J Trauma. 1997 May;42(5 Suppl):S61-5.
- Cooper et al. Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury- a randomized controlled trial.JAMA. 2004 Mar 17;291(11):1350-7.
- Bulger E, et al. Out-of-Hospital Hypertonic Resuscitation Following Severe Traumatic Brain Injury. A Randomized Controlled Trial. JAMA. 2010 Oct 6;304(13):1455-64.
- Davis D, et al. Both Hypoxemia and Extreme Hyperoxemia May Be Detrimental in Patients with Severe Traumatic Brain Injury. J Neurotrauma.2009 Dec;26(12):2217-23.
- Rincon F, et al. Significance of arterial hyperoxia and relationship with case fatality in traumatic brain injury: a multicentre cohort study, J Neurol Neurosurg Psychiatry 2013: 30; 550-5
- Davis D, et al. Early ventilation and outcome in patients with moderate to severe traumatic brain injury. Crit Care Med. 2006 Apr;34(4):1202-8.
- Dumont T, et al. Inappropriate Prehospital Ventilation in Severe Traumatic Brain Injury Increases In-Hospital Mortality.J Neurotrauma. 2010 Jul;27(7):1233-41.
- Warner KJ, et al. The Impact of Prehospital Ventilation on Outcomes After Severe Traumatic Brain Injury. J Trauma. 2007 Jun;62(6):1330-6; discussion 1336-8.
- Warner KJ, et al. Emergency Department Ventilation Effects Outcome in Severe Traumatic Brain Injury. J Trauma. 2008 Feb;64(2):341-7
- Lee Sung-Woo, et al. Concordance of End-Tidal Carbon Dioxide and Arterial Carbon Dioxide in Severe Traumatic Brain injury. J Trauma. 2009 Sep;67(3):526-30
- Berry C, et al. Redefining hypotension in traumatic brain injury. Injury. 2012 Nov;43(11):1833-7
- Ko Sang-Bae. Multimodality Monitoring in the Neurointensive Care Unit. J Stroke. 2013 May;15(2):99-108




